1.本发明涉及色料材料领域,尤其涉及一种硅铁红色料的制备方法。
背景技术:
2.硅铁红色料是一种以sio2作为包裹相,对着色剂fe2o3进行包裹的包裹色料,其在苛刻的使用环境下能稳定呈色,且颜色鲜艳,耐高温,常用作陶瓷色料。
3.制备硅铁红色料常用的方法有高温固相法和液相法,其中,液相法制备的产品包裹率、粒径、纯度和呈色性能等优于固相法,是目前制备硅铁红包裹色料最常用的方法。但是传统液相法中仍然面临所制备的硅铁红色料粒径分布宽、形态不均一,包裹不完整的缺点,从而直接影响其发色。
技术实现要素:
4.本发明的目的在于提供一种硅铁红色料的制备方法,本发明提供的方法,制备的硅铁红色料粒径分布窄、形态均一,包裹完整,发色稳定。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种硅铁红色料的制备方法,包括以下步骤:
7.(1)将咪唑类离子液体、铁源、矿化剂和水混合,得到混合溶液;
8.(2)将所述步骤(1)得到的混合溶液与正硅酸乙酯和醇混合后调节ph值,得到前驱液;所述前驱液的ph值为2~6;
9.(3)将所述步骤(2)得到的前驱液在微波加热条件下进行水热反应,得到硅铁红色料。
10.优选地,所述步骤(1)中的咪唑类离子液体包括1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐、溴化1
‑
丁基
‑3‑
甲基咪唑和溴化1
‑
辛基
‑3‑
甲基咪唑中的一种或多种。
11.优选地,所述步骤(1)中的铁源包括六水氯化铁、九水硝酸铁和水合硫酸铁中的一种或多种。
12.优选地,所述步骤(1)中的矿化剂包括氟化钠、氟化钾和氟化铵中的一种或多种。
13.优选地,所述步骤(1)中的咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比为(0.004~0.01)mol:(0.2~0.7)g:(0.10~0.23)g。
14.优选地,所述步骤(1)中的咪唑类离子液体和所述步骤(2)中的正硅酸乙酯的物质的量之比为1:(0.20~0.40)。
15.优选地,所述步骤(2)中调节ph值所用的碱为氢氧化钠或氢氧化钾。
16.优选地,所述步骤(2)中前驱液的ph值为3~5。
17.优选地,所述步骤(3)中水热反应的温度为160~230℃,水热反应的时间为1~4h。
18.优选地,所述步骤(3)中水热反应完成后还包括:将所述水热反应的产物依次进行固液分离、洗涤和干燥。
19.本发明提供了一种硅铁红色料的制备方法,利用铁源水解产生铁的氢氧化物,然
后在微波加热的反应体系中,脱水生成fe2o3纳米晶,随后正硅酸乙酯水解形成的二氧化硅包覆在所述fe2o3纳米晶表面,形成硅铁红色料,本发明利用咪唑类离子液体作为助剂,与铁源中的铁离子的配位,首先咪唑类离子液体起到“软模板剂”的作用,能够诱导fe2o3规则结晶,有利于形成粒径分布窄、形态均一的fe2o3纳米晶,其次,咪唑类离子液体可以用作“波导剂”可以高效吸收微波的能量,短时间内使得体系快速升温,从而导致fe2o3大量快速成核,消耗铁源,并控制fe2o3后期生长速率,使其缓慢生长,避免出现“奥斯特瓦尔德熟化效应”而造成fe2o3纳米晶尺寸分布不均,再者,咪唑类离子液体也可以作为“晶面抑制剂”,有效抑制fe2o3晶面异常生长,也能够改善fe2o3结晶形态均匀性,通过矿化剂协同微波水热的合成反应条件在较短时间内改善反应体系中各物质在反应体系中的迁徙,促进fe2o3成核,并控制其后期生长速率,使其缓慢长大,有利于获得尺寸分布均匀的fe2o3纳米晶,从而有利于后续制备得到粒径分布窄、形态均一的硅铁红色料,并且fe2o3纳米晶的尺寸规整均匀为后期sio2完整高效率的包覆提供了基础,通过调节前驱液的ph值(或反应体系的ph值),以控制正硅酸乙酯的水解为二氧化硅的速率,在形成fe2o3纳米晶后水解得到的二氧化硅对其进行包覆,以有利于包裹完整,最终获得粒径分布窄、形态均一的包裹完整、发色稳定的硅铁红色料。实施例的结果显示,利用本发明提供的方法,成功制备得到了硅铁红色料,所述色料的内核被包裹物为fe2o3,fe2o3纳米晶颗粒以杆状或球状被sio2很好的完整包裹,包裹状态佳,且硅铁红色料的粒径均匀分布,呈色效果较好,发色稳定,发色性能:l
*
=55.28,a
*
=22.23,b
*
=20.66,或l
*
=39.59,a
*
=33.30,b
*
=24.32,或l
*
=45.14,a
*
=25.12,b
*
=17.4。
20.本发明提供的硅铁红色料的制备方法操作简单,适宜规模化生产。
附图说明
21.图1为本发明实施例1制备硅铁红色料的工艺流程图;
22.图2为本发明实施例1制备的硅铁红色料的tem图;
23.图3为本发明实施例1制备的硅铁红色料的xrd图;
24.图4为本发明实施例2制备的硅铁红色料的tem图;
25.图5为本发明实施例2制备的硅铁红色料的xrd图;
26.图6为本发明实施例3制备的硅铁红色料的tem图;
27.图7为本发明实施例3制备的硅铁红色料的xrd图。
具体实施方式
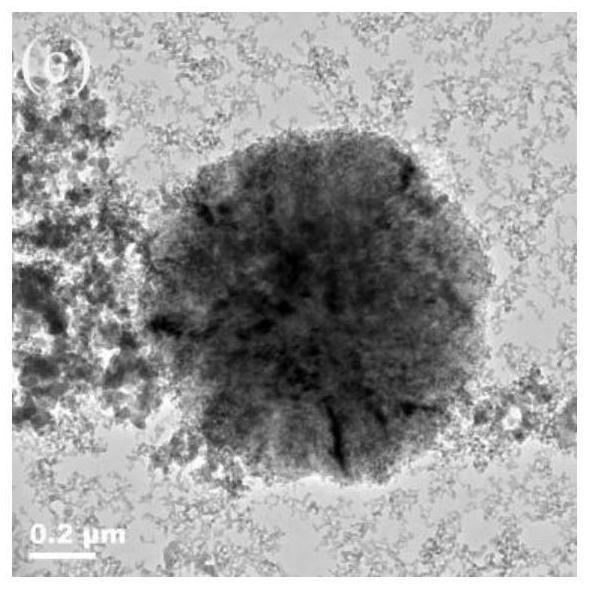
28.本发明提供了一种硅铁红色料的制备方法,包括以下步骤:
29.(1)将咪唑类离子液体、铁源、矿化剂和水混合,得到混合溶液;
30.(2)将所述步骤(1)得到的混合溶液与正硅酸乙酯和醇混合后调节ph值,得到前驱液;所述前驱液的ph值为2~6;
31.(3)将所述步骤(2)得到的前驱液在微波加热条件下进行水热反应,得到硅铁红色料。
32.在本发明中,若无特殊说明,所采用的原料均为本领域常规市售产品。
33.在本发明中,若无特殊说明,所进行的操作均为室温条件。
34.本发明将咪唑类离子液体、铁源、矿化剂和水混合,得到混合溶液。
35.在本发明中,所述咪唑类离子液体优选包括1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐、溴化1
‑
丁基
‑3‑
甲基咪唑和溴化1
‑
辛基
‑3‑
甲基咪唑中的一种或多种。
36.在本发明中,所述铁源优选包括六水氯化铁、九水硝酸铁和水合硫酸铁中的一种或多种。
37.在本发明中,所述矿化剂优选包括氟化钠、氟化钾和氟化铵中的一种或多种。
38.在本发明中,所述咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比优选为(0.004~0.01)mol:(0.2~0.7)g:(0.10~0.23)g,更优选为(0.0048~0.008)mol:(0.4~0.65)g:(0.11~0.22)g。本发明将咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比控制在上述范围内,有利于咪唑类离子液体和矿化剂充分发挥各自的作用,使fe2o3大量快速成核,有效抑制fe2o3晶面异常生长,规则结晶,并控制fe2o3后期生长速率,使其缓慢生长,获得尺寸分布均匀的fe2o3纳米晶,后续水解得到的二氧化硅对其进行包覆完整,最终获得粒径分布窄、形态均一的包裹完整、发色稳定的硅铁红色料。
39.在本发明中,所述咪唑类离子液体、铁源、矿化剂和水的混合优选包括将咪唑类离子液体和水混合,得到咪唑类离子液体溶液;将所述咪唑类离子液体与铁源和矿化剂混合,然后再搅拌,得到混合溶液。
40.在本发明中,所述搅拌的时间优选为0.5~3h,更优选为1~2.5h。本发明将搅拌的时间控制在上述范围内,有利于保证各组分完全溶解。本发明对于所述搅拌的速率没有特殊的限定,常规搅拌速率即可。
41.得到混合溶液后,本发明优选将所述混合溶液与正硅酸乙酯和醇混合后调节ph值,得到前驱液。
42.在本发明中,所述咪唑类离子液体和正硅酸乙酯的物质的量之比优选为1:(0.20~0.40),更优选为1:(0.23~0.38),进一步优选为1:(0.25~0.33)。本发明将咪唑类离子液体和正硅酸乙酯的物质的量之比控制在上述范围内,有利于后续生成的fe2o3纳米晶均匀成核,生长速率可控,及后期包覆,避免二者的物质的量之比过大,咪唑类离子液体量过多,所述反应体系的微波导热性增强,fe2o3纳米晶容易异常长大,尺寸分布不均匀,而造成后期被sio2包覆不完整,制备的硅铁红色料发色不稳定,同时避免二者的物质的量之比过小,咪唑类离子液体量过少,反应体系中h2o的量过大,导热性降低的同时,咪唑类离子液体作为“软膜板剂”及“晶面抑制剂”的作用同时降低,加之h2o在微波短时间加热后造成反应体系压力增大,最终会造成fe2o3纳米晶异常长大,尺寸分布不均匀,而造成后期被sio2包覆不完整,制备的硅铁红色料发色不稳定。
43.在本发明中,所述混合溶液与正硅酸乙酯和醇的混合优选包括将所述混合溶液与正硅酸乙酯和醇混合,得到的混合体系;将所述混合体系进行搅拌。在本发明中,所述搅拌的时间时间优选为3~40min,更优选为5~30min。本发明将搅拌的时间控制在上述范围内,有利于保证各组分完全溶解,并混合均匀。本发明对于所述搅拌的速率没有特殊的限定,常规搅拌速率即可。
44.在本发明中,所述调节ph值优选在搅拌的条件下进行。本发明对于所述搅拌的速率没有特殊的限定,常规搅拌速率即可。
45.在本发明中,所述调节ph值所用的碱为氢氧化钠或氢氧化钾。在本发明中,所述碱优选以水溶液的形式加入;所述水溶液的浓度优选为1.5~4mol/l,更优选为2~3mol/l。
46.调节ph值完成后,本发明优选将所述调节ph值完成后得到溶液进行搅拌,得到前驱液。
47.在本发明中,所述搅拌的时间时间优选为20~80min,更优选为30~60min。本发明将搅拌的时间控制在上述范围内,有利于保证各组分完全溶解,并混合均匀。本发明对于所述搅拌的速率没有特殊的限定,常规搅拌速率即可。
48.在本发明中,所述前驱液的ph值为2~6,更优选为3~5。本发明将前驱液的ph值控制在上述范围内,有利于控制正硅酸乙酯的水解为二氧化硅的速率,在形成fe2o3纳米晶后水解得到的二氧化硅对其进行包覆,以有利于包裹完整,最终获得粒径分布窄、形态均一的包裹完整、发色稳定的硅铁红色料。
49.得到前驱液后,本技术将所述前驱液在微波加热条件下进行水热反应,得到硅铁红色料。
50.在本发明中,所述水热反应的设备优选为微波水热反应釜。本发明对所述微波水热反应釜没有特殊的限制,采用本领域熟知的市售产品即可。在本发明中,所述微波水热反应釜的填充度优选为40~70%,更优选为50~60%。本发明将微波水热反应釜的填充度控制在上述范围内,有利于可以保证反应体系压力保持在一定反应,促进反应快速正向进行,诱导fe2o3纳米晶快速成核,后期结晶生长发育完整,发色饱满,同时避免填充度过低而导致fe2o3纳米晶尺寸分布不均匀,后期sio2无法高效完整包裹,导致制备的硅铁红色料在坯体中热处理后发色不稳定。
51.在本发明中,所述水热反应的温度优选为160~230℃,更优选为180~220℃。在本发明中,所述水热反应的时间为1~4h,更优选为1.5~3h。本发明将水热反应的温度和时间控制在上述范围,有利于控制正硅酸乙酯水解速率,控制sio2成核生长,完成对fe2o3纳米晶色料的包裹。
52.在本发明中,所述水热反应完成后还包括:将所述水热反应的产物依次进行固液分离、洗涤和干燥。
53.本发明对于所述固液分离的方式没有特殊的限定,采用本领域技术人员熟知的方式即可。在本发明中,所述洗涤优选包括依次进行的第一洗涤和第二洗涤;所述第一洗涤所用溶剂优选为去离子水,所述第二洗涤所用溶剂优选为无水乙醇;所述第一洗涤和第二洗涤的次数优选独立地为1~3次。在本发明中,所述干燥的时间优选为10~16h,更优选为12~14h。
54.本发明提供的硅铁红色料的制备方法操作简单,反应条件温和,适宜规模化生产,制备的硅铁红色料粒径分布窄、形态均一,包裹完整,发色稳定。
55.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
56.实施例1
57.(1)将0.00486mol的咪唑类离子液体1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐和25ml的去离子水混合,得到1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐溶液,将所述1
‑
丁基
‑3‑
甲基咪唑四氟硼酸盐溶液与0.5g铁源六水氯化铁和0.12g矿化剂naf混合,常温搅拌2h,得到混合溶液;所述
咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比为0.00486mol:0.5g:0.12g;
58.(2)将所述步骤(1)得到的混合溶液与3.3ml(0.015mol)的正硅酸乙酯和10ml乙醇混合后,常温搅拌10min,然后在搅拌条件下用3mol/l的氢氧化钠调节ph值,继续搅拌30min,得到前驱液;所述前驱液的ph值为5;
59.所述步骤(1)中的咪唑类离子液体和所述步骤(2)中的正硅酸乙酯的物质的量之比为1:0.30;
60.(3)将所述步骤(2)得到的前驱液转移到微波水热反应釜中,控制所述微波水热反应釜的填充度为50%,在微波加热条件、210℃下进行水热反应1.5h,待反应釜冷却后,固液分离收集产物,依次经去离子水和无水乙醇分别洗涤3次后,置于电热鼓风干燥箱内60℃干燥12h,得到硅铁红色料;利用北京康光光电子联合公司生产的sp
‑
1000型色度仪来对上述硅铁红色料的呈色特征和色度参数进行表征,测试条件:2
°
观察者,波长间隔20nm,照明体d65;所述硅铁红色料的发色性能:l
*
=55.28,a
*
=22.23,b
*
=20.66。
61.图1为实施例1制备的硅铁红色料的工艺流程图;
62.图2为实施例1制备的硅铁红色料的tem图,由图2可知,实施例1制备的硅铁红色料中,fe2o3纳米晶以杆状包裹在sio2中,包裹完整;
63.图3为实施例1制备的硅铁红色料的xrd图,由图3可知,标准pdf卡片编号为#33
‑
0664,黑色棱形标识的衍射峰对应fe2o3的特征峰,说明实施例1制备的色料的内核被包裹物为fe2o3,实施例1成功制备得到了硅铁红色料。
64.实施例2
65.(1)将0.00672mol的咪唑类离子液体溴化1
‑
丁基
‑3‑
甲基咪唑和25ml的去离子水混合,得到溴化1
‑
丁基
‑3‑
甲基咪唑溶液,将所述溴化1
‑
丁基
‑3‑
甲基咪唑溶液与0.46g铁源九水硝酸铁和0.20g矿化剂kf混合,常温搅拌2h,得到混合溶液;所述咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比为0.00672mol:0.49g:0.20g;
66.(2)将所述步骤(1)得到的混合溶液与4.0ml(0.018mol)的正硅酸乙酯和10ml乙醇混合后,常温搅拌20min,然后在搅拌条件下用3mol/l的氢氧化钠调节ph值,继续搅拌30min,得到前驱液;所述前驱液的ph值为3;
67.所述步骤(1)中的咪唑类离子液体和所述步骤(2)中的正硅酸乙酯的物质的量之比为1:0.28;
68.(3)将所述步骤(2)得到的前驱液转移到微波水热反应釜中,控制所述微波水热反应釜的填充度为50%,在微波加热条件、200℃下进行水热反应3h,待反应釜冷却后,固液分离收集产物,依次经去离子水和无水乙醇分别洗涤3次后,置于电热鼓风干燥箱内60℃干燥12h,得到硅铁红色料;利用北京康光光电子联合公司生产的sp
‑
1000型色度仪来对上述硅铁红色料的呈色特征和色度参数进行表征,测试条件:2
°
观察者,波长间隔20nm,照明体d65;所述硅铁红色料的发色性能:l*=39.59,a*=33.30,b*=24.32。
69.图4为实施例2制备的硅铁红色料的tem图,由图4可知,实施例2制备的硅铁红色料中,fe2o3纳米晶尺寸分布均匀,且fe2o3纳米晶以类球状包裹在sio2中,包裹完整;
70.图5为实施例2制备的硅铁红色料的xrd图,由图5可知,标准pdf卡片编号为#33
‑
0664,黑色棱形标注的衍射峰对应fe2o3的特征峰,说明实施例2制备的色料的内核被包裹物为fe2o3,实施例2成功制备得到了硅铁红色料。
71.实施例3
72.(1)将0.00531mol的咪唑类离子液体溴化1
‑
辛基
‑3‑
甲基咪唑和25ml的去离子水混合,得到溴化1
‑
丁基
‑3‑
甲基咪唑溶液,将所述溴化1
‑
丁基
‑3‑
甲基咪唑溶液与0.60g铁源水合硫酸铁和0.21g矿化剂naf混合,常温搅拌2h,得到混合溶液;所述咪唑类离子液体的物质的量、铁源的质量和矿化剂的质量之比为0.00531mol:0.6g:0.21g;
73.(2)将所述步骤(1)得到的混合溶液与3.8ml(0.017mol)的正硅酸乙酯和10ml乙醇混合后,常温搅拌25min,然后在搅拌条件下用3mol/l的氢氧化钠调节ph值,继续搅拌30min,得到前驱液;所述前驱液的ph值为4;
74.所述步骤(1)中的咪唑类离子液体和所述步骤(2)中的正硅酸乙酯的物质的量之比为1:0.33;
75.(3)将所述步骤(2)得到的前驱液转移到微波水热反应釜中,控制所述微波水热反应釜的填充度为60%,在微波加热条件、210℃下进行水热反应3h,待反应釜冷却后,固液分离收集产物,依次经去离子水和无水乙醇分别洗涤3次后,置于电热鼓风干燥箱内60℃干燥12h,得到硅铁红色料;利用北京康光光电子联合公司生产的sp
‑
1000型色度仪来对上述硅铁红色料的呈色特征和色度参数进行表征,测试条件:2
°
观察者,波长间隔20nm,照明体d65;所述硅铁红色料的发色性能:l
*
=45.14,a
*
=25.12,b
*
=17.4。
76.图6为实施例3制备的硅铁红色料的tem图,由图6可知,实施例3制备的硅铁红色料中,fe2o3纳米晶尺寸分布均匀,且fe2o3纳米晶以球状包裹在sio2中,包裹完整;
77.图7为实施例3制备的硅铁红色料的xrd图,由图7可知,标准pdf卡片编号为#33
‑
0664,黑色棱形标注的衍射峰对应fe2o3的特征峰,说明实施例3制备的色料的内核被包裹物为fe2o3,实施例3成功制备得到了硅铁红色料。
78.由实施例和附图可知,利用本发明提供的方法,成功制备得到了硅铁红色料,所述色料的内核被包裹物为fe2o3,fe2o3纳米晶颗粒以杆状或球状被sio2很好的完整包裹,包裹状态佳,且硅铁红色料的粒径均匀分布,呈色效果较好,发色稳定,发色性能:l
*
=55.28,a
*
=22.23,b
*
=20.66,或l
*
=39.59,a
*
=33.30,b
*
=24.32,或l
*
=45.14,a
*
=25.12,b
*
=17.4。本发明提供的硅铁红色料的制备方法操作简单,反应条件温和,适宜规模化生产,制备的硅铁红色料粒径分布窄、形态均一,包裹完整,发色稳定。
79.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。